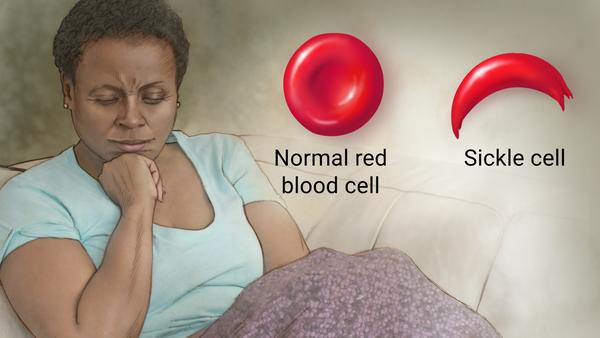
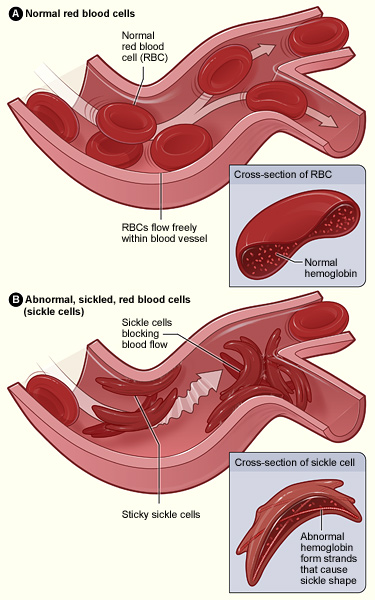
Management of sickle cell anemia is usually aimed at avoiding pain episodes, relieving symptoms, and preventing complications. Treatments might include medications and blood transfusions. For some children and teenagers, a stem cell transplant might cure the disease.
Medications
Hydroxyurea (Droxia, Hydrea, Siklos). Daily hydroxyurea reduces the frequency of painful crises and might reduce the need for blood transfusions and hospitalizations. But it can increase the risk of infections. Don’t take the drug if you’re pregnant.
L-glutamine oral powder (Endari). The FDA recently approved this drug for treatment of sickle cell anemia. It helps in reducing the frequency of pain crises.
Crizanlizumab (Adakveo). This drug, given by injection, can help reduce the frequency of pain crises in adults and children older than 16. Side effects can include nausea, joint pain, back pain and fever.
Voxelotor (Oxbryta). This drug is used to treat sickle cell disease in adults and children older than 12. Taken orally, this drug can lower the risk of anemia and improve blood flow throughout the body. Side effects can include headache, nausea, diarrhea, fatigue, rash and fever.
Pain-relieving medications. Your doctor might prescribe narcotics to help relieve pain during sickle cell pain crises.
Preventing infections
Children with sickle cell anemia might receive penicillin between the ages of about 2 months old until at least age 5 years. Doing so helps prevent infections, such as pneumonia, which can be life-threatening to children with sickle cell anemia.
Adults who have sickle cell anemia might need to take penicillin throughout their lives if they’ve had pneumonia or surgery to remove the spleen.
Childhood vaccinations are important for preventing disease in all children. They’re even more important for children with sickle cell anemia because their infections can be severe.
Your child’s doctor should ensure that your child receives all the recommended childhood vaccinations, as well as vaccines against pneumonia, meningitis, hepatitis B and an annual flu shot. Vaccines are also important for adults with sickle cell anemia.
During the COVID 19 pandemic, people with sickle cell anemia should take extra precautions, such as staying isolated at home as much as possible and for those who are eligible, getting vaccinated.
Surgical and other procedures
Blood transfusions. These are used to treat and prevent complications, such as stroke, in people with sickle cell disease.
In a red blood cell transfusion, red blood cells are removed from a supply of donated blood, then given through a vein to a person with sickle cell anemia. This increases the number of normal red blood cells, which helps reduce symptoms and complications.
Risks include an immune response to the donor blood, which can make it hard to find future donors; infection; and excess iron buildup in your body. Because excess iron can damage your heart, liver, and other organs, you might need treatment to reduce iron levels if you undergo regular transfusions.
Stem cell transplant. Also known as bone marrow transplant, this procedure involves replacing bone marrow affected by sickle cell anemia with healthy bone marrow from a donor. The procedure usually uses a matched donor, such as a sibling, who doesn’t have sickle cell anemia.
Because of the risks associated with a bone marrow transplant, including death, the procedure is recommended only for people, usually children, who have significant symptoms and complications of sickle cell anemia. A stem cell transplant is the only known cure for sickle cell anemia.
Clinical trials are ongoing to address stem cell transplantation in adults and gene therapies.